

 833-264-6620
833-264-6620


















Cystic fibrosis is a progressive genetic disorder that affects over 30,000 people in the United States, according to the Cystic Fibrosis Foundation Patient Registry. When the disease was first reported in 1938, it was described as “mucoviscidosis” to emphasize a patient’s tendency to produce profuse amounts of mucus in the body. The abundance of mucus leads to grave complications over time, as it causes frequent lung infections and impairs lung function, including restricting a patient’s ability to breathe. As researchers attempt to better understand what causes cystic fibrosis, breakthroughs in treatment are ongoing and show promise.
Depending on the severity of the illness, this lung disease can cause extensive damage to the digestive system, respiratory system, reproductive system, and vital organs. Organ damage, infections, and breathing obstruction can even lead to death. The average life expectancy for cystic fibrosis patients is between 42 and 50 years of age. Lung complications are responsible for 80% of all cystic fibrosis deaths.
Cystic fibrosis is caused by a genetic defect of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which normally regulates the production of tears, digestive enzymes, sweat, saliva, and mucus in healthy cells. Among other errors, the mutation deletes amino acid and primary nucleotide-binding domain phenylalanine 508 (F508del) in CFTR, causing faulty cell gating and erratic channel processing across a cell’s plasma membrane. The defective genes also disrupt the passage of salts across cells. There are over 1,700 known cystic fibrosis mutations; the type of cystic fibrosis defect determines the severity of the disease.
Researchers suspect that mucus hyperproduction is not a direct cause of the defective CFTR, but is a “downstream consequence,” as changes in CFTR expression in different cell types do not always correlate with disease severity or mucus buildup. Mechanisms for mucus production, per se, appear normal in most instances; therefore, cystic fibrosis genetics continues to be the focus of illness causation.
Family history is a prime cystic fibrosis risk factor. Cystic fibrosis sufferers inherit two copies of the defective gene—one from each parent who must have at least one copy of the CF gene. Single-copy carriers of the defective gene are otherwise normal but can pass the mutation on to offspring. There is a 25% chance that two cystic fibrosis gene carriers can produce healthy, non-cystic fibrosis children who are not carriers. However, there is also a 25% chance that the offspring of two cystic fibrosis gene carriers will produce a child who will have cystic fibrosis, while there is a 50% chance the child will be a carrier who does not have cystic fibrosis.
Approximately 1 in 25 people (or 10 million Americans) are carriers of the defective CF gene. Though there are carriers among all races, Caucasians of Northern European descent are most at risk of developing cystic fibrosis. The disorder is least common among Asian and African groups.
Cystic fibrosis can yield a variety of symptoms. The severity of these symptoms varies among cystic fibrosis sufferers. Symptoms of cystic fibrosis include:
The overproduction of sticky mucus yields many of these symptoms. Mucus is a viscoelastic substance that protects and covers the surfaces of the reproductive, gastrointestinal, and respiratory epithelial tracts. An overabundance of thick mucus, however, hinders the secretion of pancreatic digestive enzymes. If these digestive enzymes fail to reach the small intestine, the body is unable to process proteins, fats, and vitamins from foods. And mucus-filled lungs impair efficient breathing and decrease the flow of oxygen in the blood. Moist, mucus-filled lungs are breeding grounds for infectious bacteria that get trapped in respiratory airways.

The symptoms of cystic fibrosis often appear in infancy and childhood. Meconium ileus, or intestinal blockage, is a condition that occurs in 5% to 10% of newborns with cystic fibrosis. Newborn screening for cystic fibrosis is standard in the United States.
As they age, cystic fibrosis sufferers can experience more serious complications. Repeated coughing can produce blood from eroded tissue lining along respiratory airways. Chronic swelling inside the nose may trigger the formation of nasal polyps. Chronic lung infections can lead to more serious disorders, like pneumonia or bronchitis. Lung tissue that worsens over time can permanently limit function, inducing respiratory failure. Severe bouts of breathlessness can last for weeks and require hospitalization.
As severity of the disease progresses, pancreatic damage compromises insulin production and considerably increases the risk of diabetes in cystic fibrosis patients. Liver damage from mucus disruption can trigger gallstones, block the bile duct, and cause liver disease. Risk of osteoporosis, or low bone density, increases in cystic fibrosis patients. Worsening nutrient absorption over time is a major risk factor for bone loss and poor growth. Hypotension, or abnormally low blood pressure, heart arrhythmias, and extreme fatigue likely precipitate with the loss of metabolism-essential salts and minerals through sweat in cystic fibrosis patients.
Clubbing of toes and fingers are further signs of oxygen-starved tissues, nutrient deficiencies, and any number of recurring illnesses aggravated by cystic fibrosis. Additionally, over 90% of males who have cystic fibrosis experience high rates of infertility due to the absence or blockage (by mucus) of the vas deferens—the sperm duct that connects the testicles to the prostate gland by way of the urethra. Cystic fibrosis male infertility is further exacerbated if sperm cells are diminished or impaired (malformed or poorly motile) due to reproductive damage. Malnutrition, as well as increased mucus production along the cervical lining in women with cystic fibrosis, can increase the risk (by approximately 20%) of infertility or pregnancy difficulties.
There are genetic tests and blood tests available to screen for cystic fibrosis; however, genetic testing does not detect all known mutations of the disorder. Therefore, couples seeking to get pregnant may not receive accurate results regarding their carrier status for the CF gene. Luckily, advancements in postnatal screenings can diagnose this lung disease within the first month of life—which helps parents prepare for long-term care for their cystic fibrosis children before serious symptoms of CF and complications develop. Diagnostic sweat tests and genetic tests are available for babies and older patients.
Although there is no available cure for cystic fibrosis, treatment options are geared toward alleviating cystic fibrosis symptoms and complications to improve quality of life. Cystic fibrosis treatment plans are tailored to each patient’s specific circumstances, so it is important to work closely with your health care provider through the process. In addition to daily care such as a healthy diet and adequate fluid intake, treatment options to help mitigate CF affects include:
Although males with cystic fibrosis may not be able to father children naturally, they may still possess healthy sperm cells. Artificial insemination is a popular option in this case.
Innovative cystic fibrosis genetics research has focused on how particular CFTR gene failures may affect specific organ cells and their metabolic mechanisms. The United States Food and Drug Administration approved drugs known as CFTR modulators to treat the defective CFTR protein directly—one drug was approved in 2012, and the second drug in 2015. It is believed that CFTR modulators will add many years to the current cystic fibrosis life expectancy for some people.
The symptom-targeted remedy thymosin alpha 1 has shown great promise therapeutically in clinical trials. Thymosin alpha 1 is processed as a 28-amino acid fragment from prothymosin alpha, a 113-amino acid precursor. Thymosin alpha 1 repaired multiple tissue defects in mice with cystic fibrosis by reducing inflammation and increasing CFTR development, activity, and stasis.
Muscle wasting in cystic fibrosis patients reduces quality of life and survival rates. Conventional nutritional therapies have proven ineffective at curtailing the loss of muscle mass associated with cystic fibrosis. Building on previous research that shows essential amino acids can help increase muscle protein synthesis in populations prone to muscle wasting, researchers discovered that the muscle-building effects of an essential amino acid supplement outperform those of a high-quality whey protein supplement on pediatric patients with cystic fibrosis.
To learn more about how amino acids can help with cystic fibrosis, read this article.

Be the first to know about new craveable recipes and tips for living your best life.
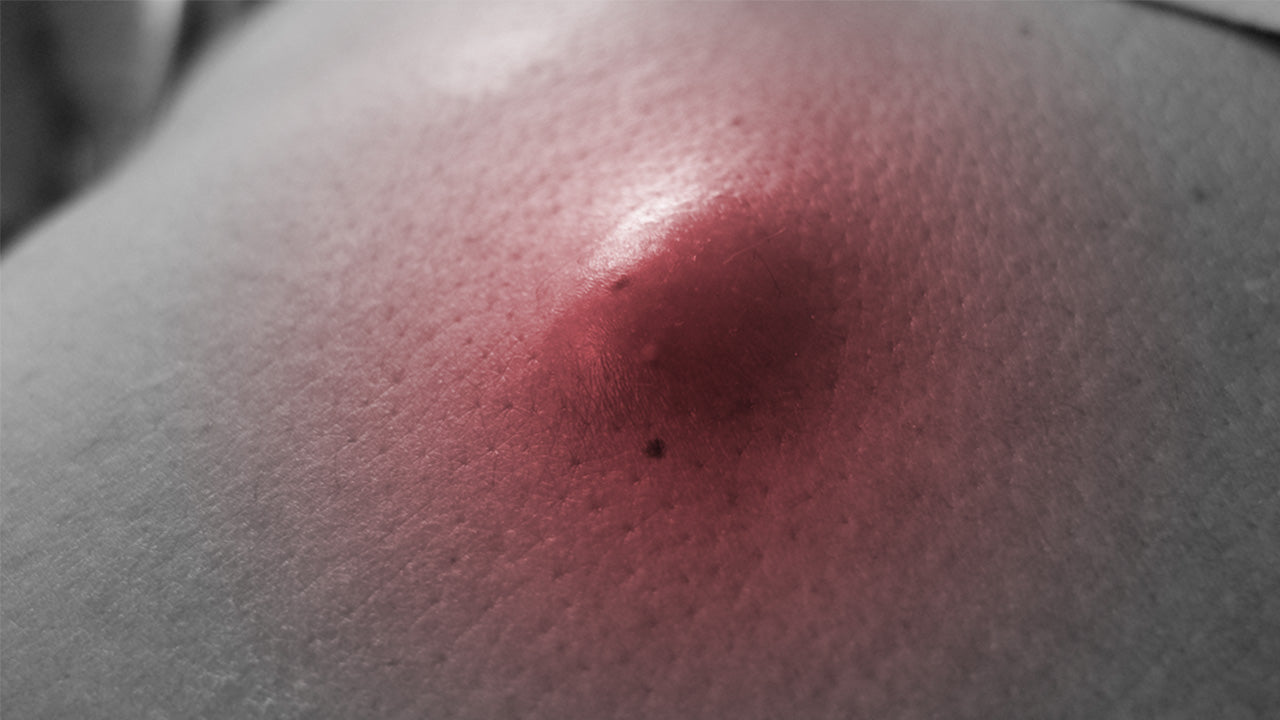
The Internet is filled with videos and pictures of people (and doctors!) popping cysts or even lancing the skin to pull out a cyst. Let’s be clear—do not pop, squeeze or lance a cyst—this is not safe. So how do you get rid of a cyst safely?

Just because ovarian cysts are benign doesn’t mean they can’t disrupt your life. So we’re here to share with you some at-home treatments to help manage ovarian cyst pain as well as share the common causes, types, and symptoms of ovarian cysts.
Sign up for our newsletter and let us know what you’re interested in, and you’ll also receive a free E-Book.
30 years of research... and still going.
Give us a try today.
If, for any reason, you don’t like us or our products, simply contact our support team within 60 days and we’ll happily refund you 100% of your payment.
It's our way of making sure you're completely happy with your purchase.
{kind=link}